
IgA-Nephropathie
Die IgA-Nephropathie (IgAN) ist weltweit die häufigste primäre Glomerulonephritisform mit einer Prävalenz von etwa 2,5/100.000 Einwohnern. Eine sehr hohe Dunkelziffer nicht diagnostizierter und vergleichsweiser benigner Fälle ist anzunehmen. Die IgAN zählt zu den Hauptursachen für Nierenversagen insbesondere bei jungen Erwachsenen bzw. zu den häufigsten Gründen für eine Nierentransplantation in dieser Altersgruppe. Männer sind zwei- bis dreimal häufiger betroffen als Frauen.
DR. MED. ANTJE HOHMANN DA SILVA
Pathophysiologisch ist die IgAN durch die Ablagerung von Immunglobulin A im glomerulären Mesangium gekennzeichnet. Dies führt zu Veränderungen der extrazellulären Matrixkomponenten, zur Proliferation von Mesangialzellen, zu einer chronischen Inflammation und schließlich zu einem Verlust von Nephronen und einer zunehmenden interstitiellen Fibrose. Bei molekularen Analysen wurde mesangiales IgA überwiegend als di- oder polymeres IgA1 nachgewiesen, wobei das IgA1-Molekül untergalaktolysiert ist („galactose-deficient IgA1, Gd-IgA1“). Dieses mesangial abgelagerte Gd-IgA1 stammt vermutlich aus Lymphozyten der intestinalen Mukosa im Sinne einer „Darm-Nieren-Achse“. Große Schwankungen in der Prävalenz lassen zusätzliche genetische als auch mögliche Umweltfaktoren in der Pathogenese vermuten.
Die meisten Diagnosen einer IgAN stellen Zufallsbefunde im Rahmen der Abklärung einer Hypertonie, Mikrohämaturie, Proteinurie oder Nierenfunktionseinschränkung dar. Die Betroffenen weisen individuell unterschiedliche Krankheitsverläufe aus, die sowohl leicht als auch rapid-progressiv ausgeprägt sein können. Mehrheitlich verläuft die Erkrankung klinisch inapparent und wird oft erst bei schon manifester Nierenfunktionsstörung mittels Nierenbiopsie diagnostiziert. Eine Spontanremission ist jedoch ebenfalls möglich.
Die Nierenbiopsie ist der Goldstandard für die Diagnose einer IgA-Nephropathie, und die initialen histopathologischen Befunde haben eine große Bedeutung für die jeweilige Prognose. Die Bewertung der Bioptate erfolgt nach dem sog. Oxford MEST-C Score, welcher fünf Parameter umfasst: mesangiale Hyperzellularität (M), endokapillare Proliferation (E), segmentale Glomerulosklerose (S), tubuläre Atrophie und interstitielle Fibrose (T) sowie glomeruläre Halbmonde (C = Crescents). Eine individuelle Prognoseeinschätzung ist bei erwachsenen Patienten über das „International IgAN Prediction Tool at biopsy“ möglich (https://qxmd.com).
Die Prognose der IgAN ist sehr variabel. Die oftmals späte Diagnosestellung ist auch auf die Tatsache zurückzuführen, dass es bisher keine spezifischen Biomarker gibt. Die Bestimmung von IgA im Serum hat keine diagnostische oder prognostische Relevanz, da die IgA-Serumkonzentration nur in etwa der Hälfte der Fälle erhöht ist. Eine persistierende Mikrohämaturie bedarf unbedingt der weiteren Abklärung. Phasenkontrastmikroskopisch werden hierbei häufig dysmorphe Erythrozyten/Akanthozyten und Erythrozytenzylinder nachgewiesen. Neben den histopathologischen Parametern der MEST-C Klassifikation sind die geschätzte glomeruläre Filtrationsrate (eGFR), der Blutdruck, das Ausmaß der Proteinurie, die Ethnie, das Alter sowie Informationen zur medikamentösen Behandlung [bspw. mit RAAS (Renin-Angiotensin-Aldosteron-System)-Inhibitoren und Immunsuppression] prognostisch bedeutsam.
Eine episodische Makrohämaturie, die häufig postinfektiös (meist nach einem viralen Infekt der oberen Atemwege) auftritt und oft mit einem akuten Nierenfunktionsverlust einhergeht, kann Anlass zur weiteren Abklärung einer möglichen IgAN sein. Die Immunglobulin A-Vaskulitis (früher Purpura Schönlein-Henoch) mit Purpura, Arthralgien, IgAN und evtl. Darmbeteiligung stellt vermutlich eine systemische Variante der IgAN dar, die besonders im Kindesalter vorkommt.
Eine sekundäre IgAN kann insbesondere bei Darmkrankheiten, Leberzirrhose und rheumatischen Erkrankungen aber auch im Zusammenhang mit verschiedenen anderen Krankheitsbildern auftreten (sieheTabelle 1).
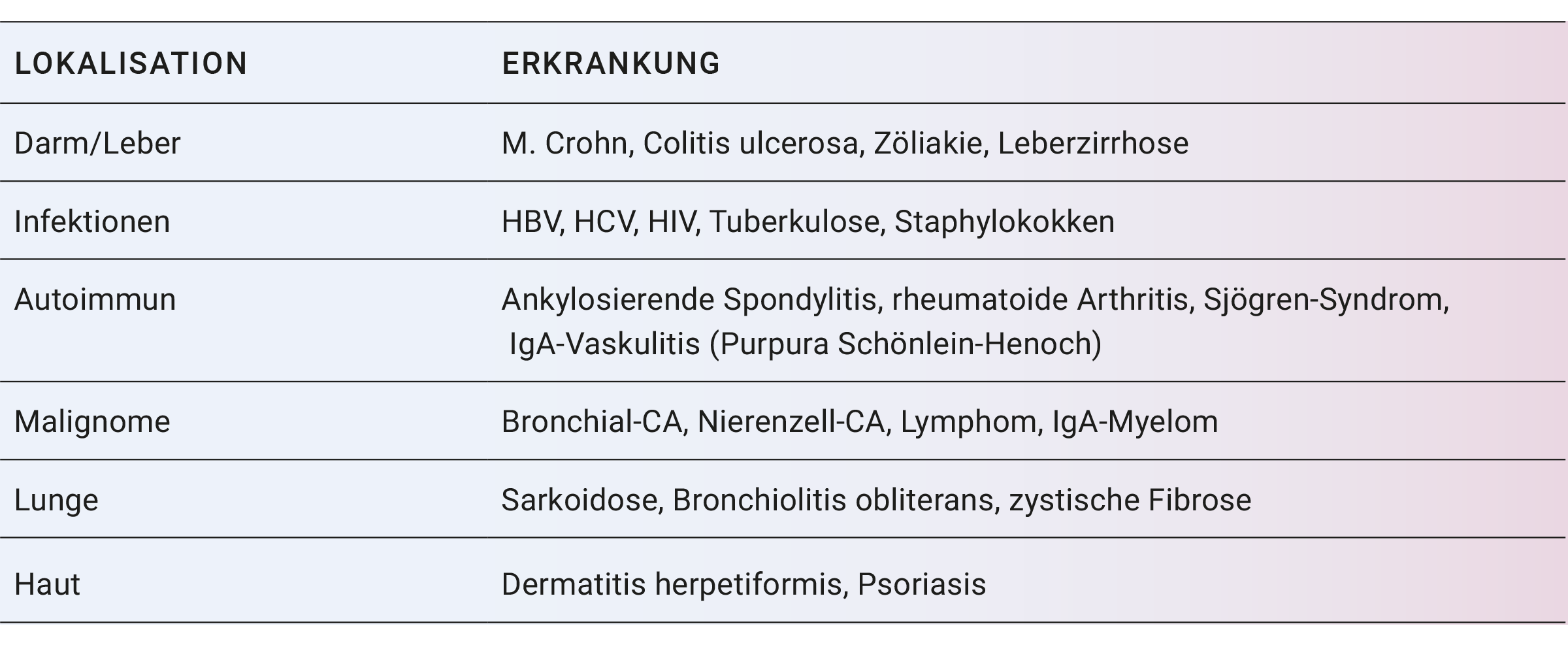
Tabelle 1. Relevante Grunderkrankungen und betroffene Organsysteme bei sekundärer IgA-Nephropathie
Angesichts des meist sehr langsamen Verlaufs der IgAN stellen umfangreiche supportive Maßnahmen zur Progressionsverzögerung einen essenziellen Grundbaustein der Behandlung dar. Diese sollen an dieser Stelle nicht weiter ausgeführt werden.
Genannt werden soll jedoch, dass der pathogenetische Hinweis auf eine zentrale Rolle des intestinalen Immunsystems und die bereits erwähnte „Darm-Nieren-Achse“ kürzlich zur Entwicklung des ersten spezifischen Medikaments zur Therapie der IgAN bei Hochrisikopatienten geführt hat. Hierbei handelt es sich um eine Budesonidgalenik, die zur präferenziellen Freisetzung von Budesonid im terminalen Ileum, dem Ort der höchste Dichte des mukosalen Immunsystems, führt.
---
Literatur
- Floege J. IgA-Nephropathie. CME Urologie 2024; 63:103–111.
- Seikrit C, Floege J. Nephrologie. https://doi.org/10.1007/s11560-024-00811-5, Springer Nature 2024, published online: 28. November 2024.
- Schimpf J et al. Diagnostik und Therapie der IgA-Nephropathie. Wien Klin Wochenschr. 2023; 135(Suppl 5):S621–S627.
- Girndt M. Immunglobulin-A-Nephropathie – neue therapeutische Möglichkeiten. Innere Medizin. 2024; 65:407–413.