
Uridindiphosphat-Glucuronyltransferase 1A1 (UGT1A1)-Testung
Die UGT1A1-Testung dient der Diagnose des Morbus Meulengracht sowie zum Ausschluss einer genetisch bedingten Irinotecan-Unverträglichkeit.
DR. MED. ATHANASIOS VERGOPOULOS
Bilirubin entsteht in erster Linie durch den Abbau des Hämoglobins. Zusätzlich wird es durch Degradierung von Muskelmyoglobin und Cytochromen gebildet. Über mehrere Zwischenstufen im reticuloendothelialen System, insbesondere der Milz, wird aus Hämoglobin zunächst indirektes (unkonjugiertes) Bilirubin gebildet. Dies wird von der Leber aufgenommen und zu direktem (konjugiertem) Bilirubin glucuronidiert und mit der Galle an den Darm abgegeben. Dort wird es weiter zu Urobilinogen und Stercobilin abgebaut. Ein Teil wird rückresorbiert, der andere Teil wird über den Darm und die Niere ausgeschieden.
Bei den genetischen (familiären) Hyperbilirubinämien kommt es im Rahmen einer Störung des Bilirubinstoffwechsels zu einer Hyperbilirubinämie. Insgesamt können die familiären Hyperbilirubinämien in zwei Gruppen eingeteilt werden. Diese Einteilung ist diagnostisch und prognostisch von Bedeutung. Das gemeinsame Leitsymptom ist ein unterschiedlich stark ausgeprägter passagerer Ikterus der Skleren und der Haut.
Die erste Gruppe ist durch die Erhöhung des unkonjugierten Bilirubins gekennzeichnet. Zu dieser Gruppe zählen der Morbus Meulengracht (auch Gilbert-Syndrom genannt) und das Crigler-Najjar-Syndrom Typ I und II. In der zweiten Gruppe zeigt sich eine Erhöhung des konjugierten Bilirubins, die zu den Krankheitsbildern Dubin-Johnson-Syndrom oder Rotor-Syndrom führt.
Morbus Meulengracht
Der Morbus Meulengracht beruht auf Gendefekten im UGT1A1-Gen, welche zu einer 60–80 %igen Reduktion der Enzymaktivität der mikrosomalen Uridin-diphosphat-Glucuronyltransferase führen. Die verminderte Enzymaktivität führt wiederum zu einer Konjugationsstörung des Bilirubins und zu einer leichten, unkonjugierten Hyperbilirubinämie.
Der Morbus Meulengracht ist das häufigste hereditäre Hyperbilirubinämie-Syndrom. Die Prävalenz von homozygoten Genträgern beträgt in der westlichen Welt zwischen 5 und 10 %. Heterozygote Träger können bis zu 30 % der Bevölkerung ausmachen. Der Erbgang ist autosomal-rezessiv.
Die Symptome treten insbesondere bei körperlichem oder psychischem Stress, bei verminderter Nahrungsaufnahme oder vermehrtem Alkoholkonsum auf. Neben häufig asymptomatischen Verläufen berichten Betroffene gelegentlich von Kopfschmerzen und Müdigkeit oder klagen über uncharakteristische abdominelle oder dyspeptische Beschwerden.
Die Hyperbilirubinämie kann zu einem unterschiedlich stark ausgeprägten Ikterus führen. Meist handelt es sich um nur einen milden Sklerenikterus, manchmal ist aber auch ein generalisierter Ikterus möglich.
Labordiagnostik des Morbus Meulengracht
Typischerweise ist das indirekte Bilirubin erhöht, jedoch selten höher als 6 mg/dl. Das direkte Bilirubin sowie die übrigen Laborwerte bleiben in der Regel normal. Zur Sicherung des Morbus Meulengracht ist heutzutage eine molekulargenetische Analyse des UGT1A1-Gens die Methode der Wahl.
Meist liegt bei Kaukasiern dem M. Meulengracht eine TA-Insertion von zwei Basen in der sogenannten TATA-Box der Promotorregion zugrunde. Dieses Allel wird als UGT1A1*28 oder „[TA]7-Allel“ bezeichnet und weist 7 TA-Repeats auf, im Gegensatz zum Wildtyp-Allel (UGT1A1*1 oder „[TA]6-Allel“) (siehe Abbildung 1). Kommt es zu dieser Insertion von zwei Basen in der TATA-Box, so wird das Gen vermindert exprimiert und die Enzymaktivität wird herabgesetzt. In Einzelfällen wurden auch die Insertion von vier Basen sowie die Deletion von zwei Basen beschrieben, wobei letztere mit einer Erhöhung der Enzymaktivität assoziiert ist.
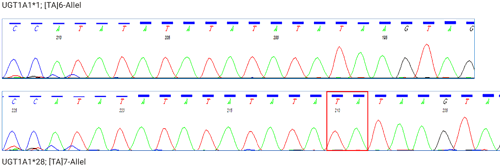 Abbildung 1. Ausschnitt eines Sequenzierchromatogramms des UGT1A1-Promotors
Abbildung 1. Ausschnitt eines Sequenzierchromatogramms des UGT1A1-Promotors
In Asien sind die Veränderungen in der Promotorregion hingegen selten. Hier wird häufig eine Mutation in Exon 1 gefunden, die zu einem Austausch von Glycin gegen Arginin in Codon 71 führt. Die Allelhäufigkeit für dieses Defizienz-Allel (UGT1A1*6) liegt beispielsweise in China und Korea bei 23 %.
Verstoffwechslung von Irinotecan
Der UGT1A1-Genlocus ist nicht nur für die Kopplung von Bilirubin an Glucuronsäure wichtig sondern auch für die Glucuronidierung von Irinotecan-haltigen Medikamenten. Irinotecan ist ein Zytostatikum und wird im Rahmen der Chemotherapie unter anderem zur Behandlung von Darmkrebs und Pankreaskarzinom eingesetzt.
Irinotecan ist ein Prodrug, das durch Carboxylesterasen in Leber und Blut zum aktiven Metaboliten SN-38 verstoffwechselt wird. SN-38 ist ein Inhibitor der Topoisomerase I und damit der DNA-Replikation. SN-38 wird zum Abbau in der Leber an Glucuronsäure gekoppelt. Die UGT1A1-Enzymaktivität spielt dabei die bedeutendste Rolle. Der Abbau und die Inaktivierung des toxischen SN-38 ist also durch Mutationen im UGT1A1-Gen und die resultierende herabgesetzte Enzymaktivität beeinträchtigt. Irinotecan kann dann zu starken Toxizitäten mit ausgeprägter Diarrhö oder Panzytopenie führen.
Empfehlungen zur UGT1A1-Testung vor Irinotecan-Gabe
Das Bundesinstitut für Arzneimittel und Medizinprodukte empfiehlt, vor Beginn einer systemischen Therapie mit Irinotecan-haltigen Arzneimitteln eine UGT1A1-Genotypisierung durchzuführen. Im entsprechenden Rote-Hand-Brief von Dezember 2021 zu Irinotecan-haltigen Arzneimitteln wird darauf hingewiesen, dass eine geringere Irinotecan-Anfangsdosis bei Patienten mit verringerter UGT1A1-Aktivität in Betracht gezogen werden sollte. Dies gilt für Patienten, denen Dosen von über 180 mg/m² Körperoberfläche verabreicht werden, oder die geschwächt sind.
Der EBM fordert bei der UGT1A1-Genotypisierung obligat die Untersuchung der Allele UGT1A1*28 und UGTA1*6. Seit Oktober 2023 wird diese molekulargenetische Analyse in unserem Labor durchgeführt.
Vom Genotyp zur Dosis
Bei Homozygotie für das UGT1A1*28- bzw. das UGT1A1*6-Allel oder Compoundheterozygotie für beide Allele, sollte eine Reduktion der Anfangsdosis erwogen werden. Die Dosis kann in den nachfolgenden Zyklen je nach Verträglichkeit und Neutrophilenzahl erhöht werden. Bei heterozygoten Trägern der untersuchten Mutationen wird keine Dosisreduktion empfohlen.
Diagnostik vor Therapiebeginn
Eine pharmakogenetische Untersuchung mit dem Ziel einer Optimierung der Arzneimitteltherapie, wie im konkreten Fall, stellt eine diagnostische genetische Untersuchung dar. Die Aufklärung über diese Untersuchung muss ärztlich erfolgen, ist aber nicht an eine dezidierte genetische Beratungskompetenz, etwa die fachgebundene genetische Beratung gebunden. Wird eine klinisch relevante UGT1A1-Variante gefunden, ist dem Patienten eine genetische Beratung anzubieten.
Therapieüberwachung
Die UGT1A1-Testung kann eine Irinotecan-Toxizität nicht vollständig ausschließen, da weitere genetische und nicht-genetische Faktoren Einfluss auf die Verträglichkeit der Medikation haben können. Engmaschige Blutbildkontrollen sind daher bei der Gabe von Irinotecan-haltigen Medikamenten weiterhin unentbehrlich. UGT1A1-Inhibitoren (z. B. Atazanavir, Indinavir) erhöhen ebenfalls die Wirkung und das Risiko für unerwünschte Arzneimittelreaktionen von Irinotecan.
Diagnostik bei Verdacht auf M. Meulengracht
UGT1A1-Genotypisierung: UGT1A1*28, ggf. UGT1A1*6
EDTA-Vollblut und Einwilligung zu genetischen Untersuchungen nach GenDG
Empfohlene Diagnostik vor Therapiebeginn mit Irinotecan
UGT1A1-Genotypisierung: UGT1A1*28 und UGT1A1*6
EDTA-Vollblut und Einwilligung zu genetischen Untersuchungen nach GenDG
Diagnostik während der Therapie mit Irinotecan
Blutbildkontrollen
EDTA-Vollblut
---
Literatur:
- The Pharmacogenomics Knowledge Base (http://www.pharmgkb.org).
- Rote-Hand-Brief zu irinotecanhaltigen Arzneimitteln vom 21.12.2021
- Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie. Positionspapier Irinotecan: Wechselwirkungen und Nebenwirkungen. Oktober 2019
- All You Need to Know About UGT1A1 Genetic Testing for Patients Treated With Irinotecan: A Practitioner-Friendly Guide. JCO Oncol Pract 2022 Apr;18(4):270–277