
Immunglobulin-G4 (IgG4)-assoziierte Erkrankungen
Unter IgG4-assoziierten Erkrankungen versteht man immunvermittelte, fibrosierende Systemerkrankungen, die klinisch meist langsam progredient und nicht hochentzündlich sind. Histologisch zeichnen sie sich durch plasmazellreiche Infiltrate mit vermehrten IgG4-positiven Plasmazellen und fibrotischen Veränderungen aus. Meist finden sich erhöhte IgG4-Konzentrationen im Serum.
DR. MED. ANTJE HOHMANN DA SILVA
Ihre genaue Prävalenz ist noch unbekannt, Männer sind jedoch häufiger betroffen als Frauen. Der Altersgipfel liegt im mittleren bis höheren Lebensalter. Pathophysiologisch ist am ehesten von einer autoimmunen Genese auszugehen. Dabei ist IgG4 selbst nicht krankheitsauslösend sondern stellt eher ein Epiphänomen dar, das zytokinvermittelt auftritt. Die charakteristische Fibrose wird durch eine ebenfalls erhöhte Produktion an profibrotischen Zytokinen begünstigt.
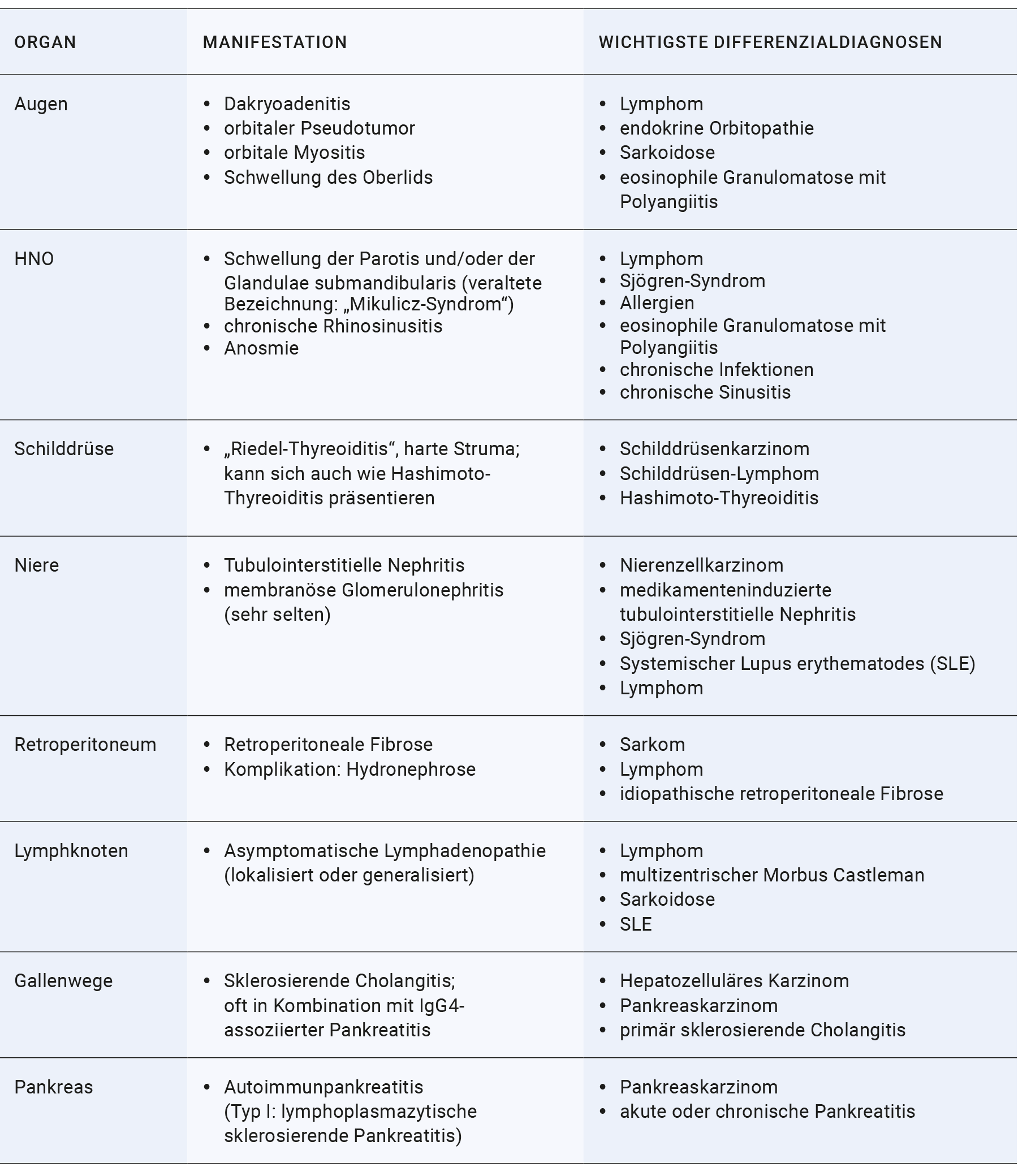
Die klinischen Manifestationen der IgG4-assoziierten Erkrankungen sind sehr variabel und können nahezu alle Organsysteme betreffen. Diese können gemeinsam oder isoliert auftreten.
Diagnose
Labordiagnostisch findet sich in den meisten Fällen ein erhöhtes Serum-IgG4, wobei der Grad der Erhöhung sehr variabel ist. Bei isolierter IgG4-assoziierter retroperitonealer Fibrose kann diese auch fehlen. Die IgG4-Konzentration fällt oft zu Beginn der immunsuppressiven Therapie ab, korreliert im Verlauf aber nicht immer mit Krankheitsschwere oder Krankheitsaktivität. Das C-reaktive Protein ist nur bei etwa der Hälfte der Patienten (selten stark) erhöht.
Bei klinischem Verdacht auf eine IgG4-assoziierte Erkrankung sollten unbedingt Biopsien der betroffenen Organe gewonnen werden. Histopathologisch
sind dichte lymphoplasmazelluläre Infiltrate mit vermehrten IgG4-positiven Plasmazellen charakteristisch. Darüber hinaus findet sich ein variables Maß an Fibrose. Die Bildgebung (Sonographie, CT, MRT, PET-CT) leistet einen guten diagnostischen Beitrag zur Diagnoseevaluation und zur Beurteilung der Ausbreitung der Erkrankung.
Zusammenfassend wird die Diagnose von IgG4-assoziierten Erkrankungen anhand von klinischen, serologischen, histologischen und bildmorphologischen Befunden gestellt, wobei zahlreiche maligne oder andere entzündliche Erkrankungen ausgeschlossen sein müssen. ACR (American College of Rheumatology) und EULAR (European League Against Rheumatism) haben im Jahr 2019 hierzu gemeinsam entsprechende Klassifikationskriterien und eine Reihe von Ausschlusskriterien definiert.
Liegen charakteristische klinische oder radiologische Befunde an typischerweise betroffenen Organen vor und wird kein Ausschlusskriterium erfüllt (von Laborseite zählen hierzu bspw. eine unklare Leukopenie und Thrombopenie, die periphere Eosinophilie, der Nachweis von ANCA, SS-A- oder SS-B-AK, AK gegen dsDNA oder anderer krankheitsspezifischer AK bzw. eine Kryoglobulinämie), dann erfolgt die Prüfung und Wichtung der einzelnen klinischen, histopathologischen und serologischen Einschlusskriterien anhand eines Scores. Labordiagnostisch wird hierfür die Serum-IgG4-Konzentrationen je nach Grad der Erhöhung mit 4 bis11 Punkten berücksichtigt, und bei Vorliegen einer Hypokomplementämie werden 6 Punkte vergeben. Bei einer Gesamtpunktzahl ≥ 20 gelten die ACR/EULAR-Klassifikationskriterien der IgG4-assoziierten Erkrankungen als erfüllt.
Symptomatische aktive Erkrankungen machen abhängig vom Schweregrad eine Behandlung erforderlich. Bei Organmanifestationen werden initial Glukokortikosteroide verabreicht. Bei Rezidiven oder schwerer Organbeteiligung kann eine längerfristige Immunsuppression indiziert sein.
Literatur
- Thiele T, Witte T. Immunglobulin-G4 (IgG4)-assoziierte Erkrankungen. Z Rheumatol 2022; 81:225–235
- Wallace ZS, Naden RP, Chari S et al. (2020). The 2019 American college of rheumatology/European league against rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis 79(1):77–87. https://doi.org/10.1136/annrheumdis-2019-216561